Download this page as a pdf by clicking here.
CONTENTS
1. What is Congenital Erythropoietic Porphyria?
2. How common is CEP?
3. What are the features of CEP?
4. How is CEP inherited?
5. How is CEP diagnosed?
6. Can CEP be diagnosed in pregnancy?
7. Is there a cure for CEP?
8. What other treatments are available for CEP?
9. Will there be new treatments available for CEP in the future?
10. Can certain medications make CEP worse?
11. What other precautions do CEP patients need to take?
12. Where can I get more information about CEP?
1. What is Congenital Erythropoietic Porphyria?
Congenital erythropoietic porphyria (CEP), also called Günther’s disease after the doctor who described it in 1911, is an inherited disease and the rarest of the main types of porphyria. In CEP, the activity of an enzyme called uroporphyrinogen III synthase (UROS) is very low. This leads to increased production of porphyrins, called type I isomer porphyrins, from the bone marrow. These porphyrins accumulate in the body, especially in the red blood cells, and cause the problems associated with CEP.2. How common is CEP?
CEP is extremely rare. As it is so rare, the exact number of people affected by CEP is not clear. It is estimated that about 1 in every 2 – 3 million people are affected by CEP. CEP can affect males and females equally, and any ethnic group.3. What are the features of CEP?
Individuals with CEP may not have all of the features described here. Different individuals may have different severity of the disease. Usually, the disease shows itself soon after birth or in early childhood, but sometimes onset of disease is delayed until adolescence or early adulthood.- Red urine is usually the first sign noticed in newborn babies with CEP. This is due to the large amount of porphyrin passed in the urine. The intensity of the redness of the urine can vary from day to day.
- The skin is very sensitive to light, especially direct sunlight or intense artificial light, such as the very bright light sometimes used to treat babies with jaundice. This causes the skin to become fragile and blister or ulcerate. This most commonly happens at sun-exposed sites, for example the backs of the hands, the face, ears and scalp. The skin may take longer to heal after injury or blistering, and become infected. Repeated blisters, wounds and ulcers can cause scarring in the skin and bald patches on the scalp.
- Some individuals may develop darkening of sun-exposed skin.
- Eyes may also be sensitive to bright sunlight or artificial light, which can cause ulcers and scarring of the eyes. With time some patients loose their eyelashes, which make their eyes prone to irritation from small particles of dust and fibres.
- Anaemia (a low haemoglobin), which varies in severity, is another feature of CEP. Anaemia develops because porphyrin damages some red blood cells which are then removed and destroyed by an organ in the abdomen called the spleen. The symptoms of anaemia include feeling tired, short of breath following minimal exertion and looking pale. A blood test will confirm the presence of anaemia.
- The spleen can gradually become bigger and cause worsening of the anaemia, and a reduction in the number of platelets (the blood cells that help to form blood clots to stop bleeding) and white cells (the blood cells that fight infections) in the blood leading to increased risk of bleeding (such as repeated nose bleeds) and infections.
- Teeth are discoloured by porphyrin causing them to appear reddish brown, especially the milk teeth.
- CEP can occasionally cause thinning of the bones (osteoporosis). Osteoporosis can lead to breakage of bones (fracture) following minimal injury.
- Excess body hair may develop, especially on the face and backs of the hands.
4. How is CEP inherited?
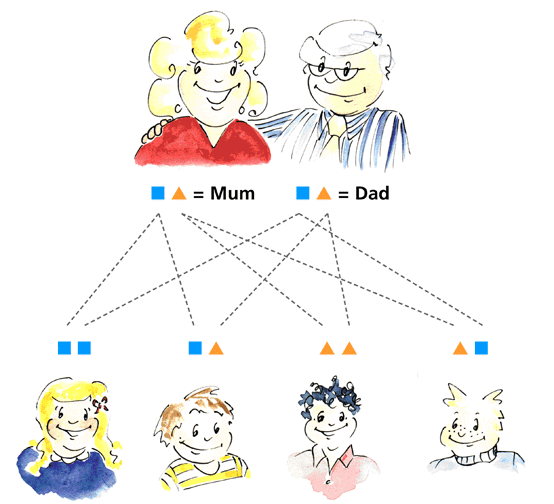
The low UROS enzyme activity in CEP is due to alterations (mutations) in the UROS gene that codes this enzyme. Each individual has 2 copies of every gene, one inherited from their mother, and one from their father. To develop CEP, one has to have two copies of the mutated gene, one inherited from each parent as shown in the diagram. This form of inheritance is called “autosomal recessive” (“autosomal” because the gene concerned is not located on the sex chromosomes). Although the parents of individuals with CEP can pass on the mutated gene to one of their children, they themselves do not have the disease, because they also have one normal gene. Similarly, some brothers or sisters of the affected person may also inherit one mutated gene from one of the parents, but because they also inherit a normal gene from the other parent, they do not have CEP. Individuals who have one mutated gene and a normal gene (such as the parents, and 2 of the children in this diagram) are called ‘carriers’ of the disease. = Normal UROS gene
= Normal UROS gene  = Mutated UROS gene
= Mutated UROS gene
|
Mum unaffected
|
Dad unaffected
|
|
Unaffected child
|
Unaffected child
|
Affected child
|
Unaffected child
|
5. How is CEP diagnosed?
CEP may be suspected in children (or rarely adults) who present with the features described above. The diagnosis is confirmed by measuring porphyrin levels in the individual’s blood, urine and faeces. These samples need to be protected from light until tested. A blood sample may also be taken to look for genetic mutations.6. Can CEP be diagnosed in pregnancy?
Testing for CEP in pregnancy is not offered routinely. However CEP can be diagnosed in pregnancy in families where there is already a child with CEP. In this situation, a test called “amniocentesis” is carried out at about 16 weeks of pregnancy. Alternatively, another test called “chorionic villus sampling” is carried out at about 12 weeks of pregnancy to collect blood cells from the placenta arising from the baby. These cells are then checked for the UROS gene mutations causing CEP.7. Is there a cure for CEP?
Currently, the only available cure for CEP is a bone marrow transplant (BMT). This involves transplanting healthy bone marrow from another person (the donor) to that of the person with CEP (the recipient). Following successful BMT, the features of CEP such as photosensitivity and anaemia will resolve. However the scarring from previous damage to the skin is permanent. For BMT to succeed, the bone marrow of the donor needs to be a good match with the recipient. BMT is a high-risk treatment where powerful treatments to suppress the recipient’s immune system are initially needed to prevent rejection. BMT is currently reserved for those severely affected individuals who have a matched bone marrow donor.8. What other treatments are available for CEP?
The treatment of CEP is aimed at preventing scarring of skin and eyes, and treatment of the complications mentioned above. Some or all of the following measures may be needed:- Protection of exposed skin from direct sunlight is required to prevent blistering and scarring. Rigorous photoprotection should include the routine use of clothing, gloves, a broad brimmed hat, scarf, long sleeves, high collars and long trousers. Conventional sunscreens (that block ultraviolet light) are not effective in CEP where the photosensitivity is to visible light. Reflectant sunscreens formulated to reflect visible light from the skin surface are required. Tinted reflectant sunscreens are available which can be mixed to match the patient’s individual skin colour. Examples of reflectant sunscreen products available on prescription and from chemists include:
- Ambre Solaire® lotion SPF 60 - RoC total Sunblock® lotion SPF 25 - Delph® lotion SPF25 - Sunsense® Ultra SPF 60 - Delph® lotion SPF 30 - Uvistat® cream SPF 22 - E45 Sun® lotion SPF25 - Ultrablock® cream SPF30 - E45 Sun® lotion SPF50 - Curtains or blinds in the house and work place may be needed to reduce the intensity of visible light. Additionally, opaque window films may be applied to the windows of buildings and/or vehicles. It is important to confirm that any window film that you select for your vehicle is legally acceptable for driving laws within your country.
- Cosmetic camouflage may be used to conceal scarring of the skin. (e.g. British Association of Skin Camouflage www.skin-camouflage.net)
- The eyes should be protected from sunlight by the use of tinted, wrap-around sunglasses. Specialist care from an ophthalmologist may be needed.
- Skin on light-exposed areas should be protected against minor trauma to prevent long-term scarring. This can be achieved by keeping the skin well moisturised and by wearing gloves.
- Skin ulcers need to be kept clean, dressed appropriately and any infections treated with antibiotic creams or tablets to speed up the healing.
- Repeated scarring of the skin, especially of the fingers, can restrict joint mobility. Regular, gentle hand exercises may help to delay or prevent this.
- Advice from an occupational therapist may be needed in patients who develop restricted hand function due to scarring of the skin.
- Blood transfusions may be needed to treat the anaemia. However, regular blood transfusions can result in iron overload. Treatment for iron overload involves a tablet or injection. Enlargement of the spleen may worsen the anaemia, necessitating removal of the spleen by an operation.
- If thinning of the bones is detected (by x-rays and bone scans), treatment with tablets may be needed.
- Good oral hygiene is important to prevent tooth decay. If opening the mouth is restricted due to scarring around the mouth, a soft children’s toothbrush or an electric toothbrush may be easier to use and cause less damage to the gums.